The putative chimeric chitinases designed by the team is a novel protein designed by orchestrating the best attributes of the wild-type chitinases. To analyze the efficiency of the designed protein structures to chitin polymer, we simulated the molecular docking process to calculate its binding affinity.
SWISS-MODEL is a web-based server specializing in homology-based protein structure modeling. It is used to generate reliable structural information of the novel chimeric protein. A lot of development has taken place in the software since its development 28 years ago. It helps users with minimal computational expertise to generate reliable protein models and easily visualize and interpret the results. As more structures are being experimentally determined, it has been noticed that the surfaces that interact with the protein's counterpart are often conserved among structures that share homology.
Raptor X is a 3D structure prediction method based on template-based modeling, homology modeling, and protein threading. This method can be implemented to make it easy to turn on and off specific features and specify which pairwise relationships should be predicted. This method works well for distantly related proteins and proteins with very few annotated sequence profiles.
We proceeded with modeling a 3-Dimensional using a fully automated protein structure homology-based modeling server namely SWISS-MODEL. Using the above-mentioned server we were successful in getting a predictive structure for our chimeric proteins but due to the fact that the predictive structures were based on homology, some parts of the protein were truncated and it showed us fragments of the final protein based on similar proteins already annotated. This led us to use another contact prediction server developed by the Xu group, excelling at tertiary and contact prediction for protein sequences without close homologs in the Protein Data Bank (PDB) called Raptor X. The RaptorX server could not provide the predicted structure of the chitinase protein from Pseudoalteromonassp DL-6. This could be due to the large nucleotide sequence of the protein. The protein structures predicted by RaptorX have been visualized using UCSF Chimera.
BC1
BC2
PC1
PC2
To find the effective binding of the chitin polymer to the chimeric chitinase protein compared to the wild-type chitinases, AutoDock software (Autodock tools-1.5.6 and AutoDock Vina) was used. It is a molecular modeling simulation software and is especially effective for protein-ligand docking. Here the ligand used was Chitin octamer (CID 24978517) since no other long-chain chitin polymer was present in the online repositories (visualised in UCSF Chimera).
Figure 1. Chitin octamer (CID 24978517)
The protein structures were prepared before docking by removing water molecules, adding hydrogen atoms, and adding Kollman charges. These modifications are necessary for the efficient binding of the ligand to the protein through non-covalent interactions.
The software was run using the structures determined by the RaptorX server and the data was as follows:
| Mode | Affinity (kcal/mol) | Dist from RMSD L.B. | Best mode RMSD U.B. |
|---|---|---|---|
1 |
-16.5 |
0 |
0 |
2 |
-15.8 |
7.224 |
14.44 |
3 |
-15.4 |
1.212 |
2.709 |
4 |
-15 |
21.146 |
26.965 |
5 |
-15 |
21.907 |
26.347 |
6 |
-14.8 |
24.365 |
27.715 |
7 |
-14.7 |
5.631 |
16.924 |
8 |
-14.6 |
6.803 |
12.991 |
9 |
-14.5 |
8.903 |
15.797 |
| Mode | Affinity (kcal/mol) | Dist from RMSD L.B. | Best mode RMSD U.B. |
|---|---|---|---|
1 |
-11.8 |
0 |
0 |
2 |
-11.7 |
1.287 |
2.467 |
3 |
-10.8 |
6.245 |
14.777 |
4 |
-10.8 |
2.900 |
10.137 |
5 |
-10.7 |
3.885 |
10.571 |
6 |
-10.6 |
3.525 |
8.198 |
7 |
-10.6 |
7.961 |
14.579 |
8 |
-10.6 |
4.779 |
10.805 |
9 |
-10.6 |
33.198 |
39.540 |
| Mode | Affinity (kcal/mol) | Dist from RMSD L.B. | Best mode RMSD U.B. |
|---|---|---|---|
1 |
-14.8 |
0 |
0 |
2 |
-14.6 |
20.789 |
30.321 |
3 |
-14.5 |
3.308 |
7.029 |
4 |
-14.3 |
2.898 |
8.011 |
5 |
-14.3 |
21.191 |
30.614 |
6 |
-14.2 |
3.355 |
7.831 |
7 |
-14 |
3.151 |
8.385 |
8 |
-14 |
2.015 |
2.781 |
9 |
-14 |
20.074 |
27.084 |
| Mode | Affinity (kcal/mol) | Dist from RMSD L.B. | Best mode RMSD U.B. |
|---|---|---|---|
1 |
-12.4 |
0 |
0 |
2 |
-12.2 |
12.275 |
21.421 |
3 |
-12.2 |
33.606 |
37.765 |
4 |
-12.0 |
9.206 |
17.650 |
5 |
-11.9 |
9.028 |
16.936 |
6 |
-11.9 |
2.270 |
3.084 |
7 |
-11.8 |
7.889 |
15.163 |
8 |
-11.8 |
1.185 |
2.287 |
9 |
-11.8 |
9.041 |
15.674 |
More the negative value of the affinity (kcal/mol), higher is the binding efficiency of the protein to the chitin polymer.
From the affinity values, it is concluded that,
To visualize the better binding of the chimeric chitinases, the binding affinity data were used to calculate its dissociation constant.
The ligand-receptor complex concentration at any given time is given by:
\[\begin{equation} \frac{d[L-R]}{dt}=k_{1}([L]_{T}-[L-R])([R]_{T}-[L-R])-k_{2}[L-R] \end{equation}\]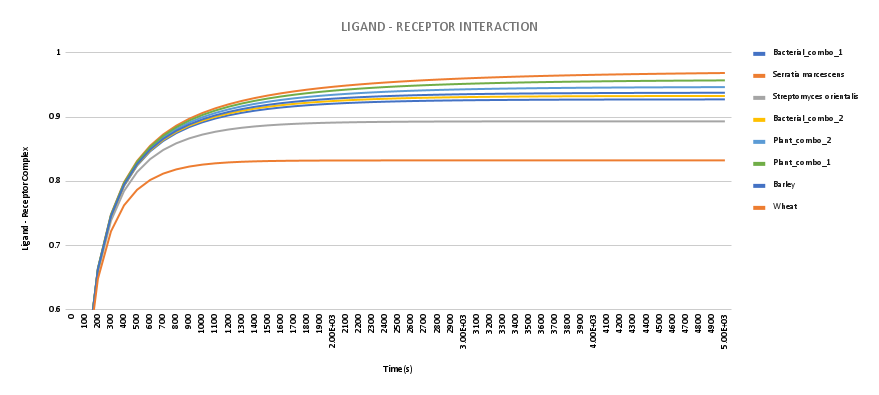
Figure. The ligand-receptor complex conc. was calculated using J-Sim v2 software.
Hence we see a higher binding affinity, therefore higher ligand-receptor concentration at any given time, with respect to wild-type chitinase proteins.